Phylogeny for the microbiologist
Being involved both in microbiology and bioinformatics I feel necessary to develop solutions to give a easy access to microbial phylogenetic reconstructions for non specialists
The "Phylogeny on the Fly" (PPF) project
"Phylogeny on the Fly" is a long-going project but we hope to publish our solution soon.
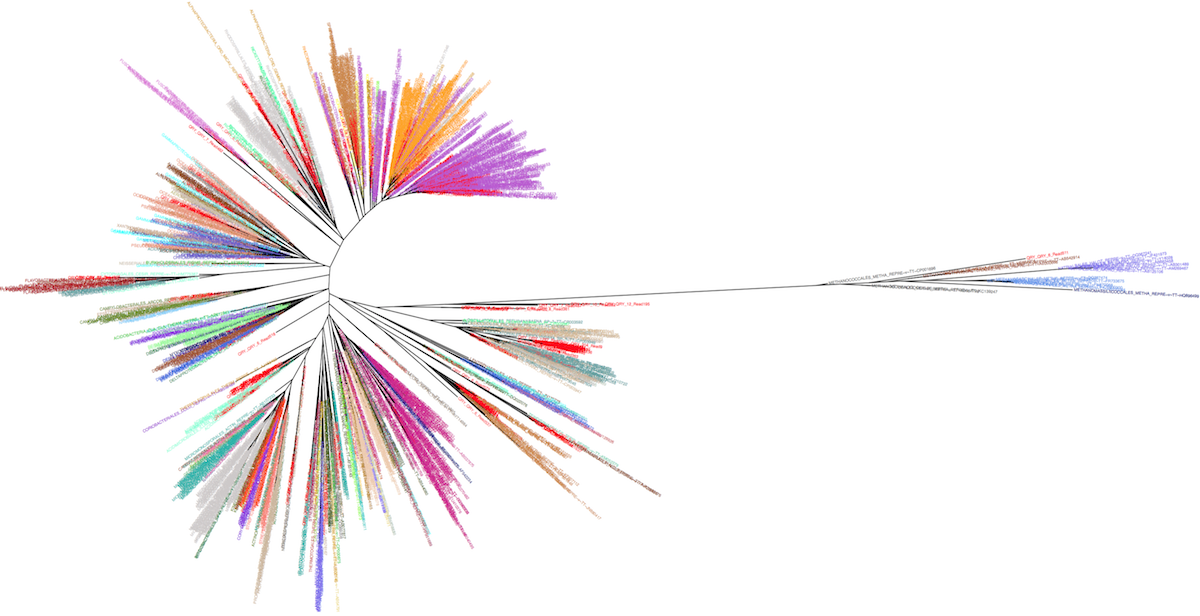
The idea is to take advantage of the leBIBI Russian doll databases to build phylogenies for a given level included in wider phylogenetic coverage without the overcrowding of non significant sequences. It is possible to extract high similarity sequences around a query at the species level or to extract sequences belonging to a given species or other levels (see the case-study of a laguna microbiome showing the position of the clusters nodes -in red- among Bacteria and Archaea).
Manolo Gouy and I have created an interactive webtool to represent the corresponding tree, enabling the visualisation of the desired taxonomic level information.
The "AmalPhy_lab" project

"AmalPhy" is not the acronym of "A Multi Agent Locus Phylogeny", even if this could have sense. The name was originaly a reference to the italian city and a joke based on a french approximate homophony. AmalPhy is not a real pipeline that take the output from a program to send to another one, it do this but with a dose of back-loops or recursivity and it construct a composite result using five different approach of the phylogenetic reconstruction for multi-locus phylogenies. Surprisingly I have been able to use many part of the PPF code and the last outputs may be viewed through PPF.
Amalphy V1 was written in 2007 for the Mycobacteria phylogeny paper. I have written the current version in 2016 to enable a direct use (without human expertise or programming) of the outputs of RiboDB (figure, output of Actinobacteria phylogeny). AmalPhy is a pipeline with a dose of back-loops and recursion. The program instructions are executed from a text pilot file that enables the iterative sending of jobs without human interaction. It accepts as input multiple families of proteins (e.g., ribosomal proteins) or a single one. In that latter case, quality-insurance protocols relying on family comparison are not possible. AmalPhy steps are a) a codon-based alignment of the sequences in a gene family, b) the selection of gene families/sequences/genomes based on content statistics, c) the sequence re-alignment and trimming (sequences in a set are aligned each time a change occurs), d) a dataset reduction by clustering (optional), e) the outliers detection and exclusion, f) the sequence alignment and concatenation, g) the concatenate translation and/or recoding (multiple schemes available), and h) the maximum likelihood-based phylogeny reconstruction (IQTree or phyML) with correction of the support values by Booster or AI-like rules.
Amalphy has been also successfully used to produce phylogenies of core genome proteins. I am open to cooperation with colleagues who want to use RiboDB (or other) to build multi-locus phylogenies.