Ribo-DB
The Ribo-DB project is one of the flagship projects of our team, it involves four researchers, 2 engineers and one doctoral student.
The ribosomal proteins are increasingly used because their number (90 proteins), the low probability of horizontal transfer, their scalable features make it an indispensable tools for the study of the evolution of organisms (phylogeny to large scale or short evolutionary scale). Furthermore, these proteins are used in the identification of pathogenic bacteria with MALDI-TOF mass-spectrometry and are also potential targets for PCR amplication in diagnostic tools.
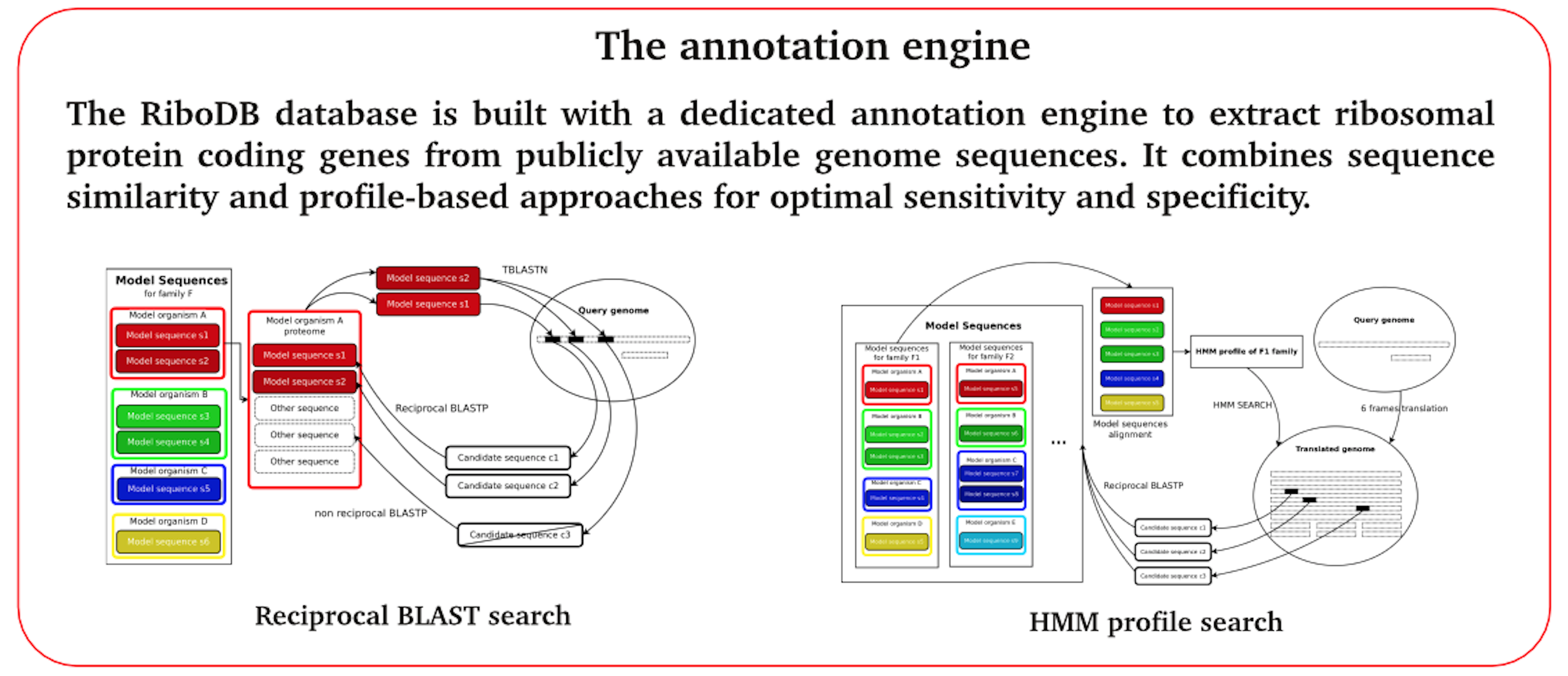
No basic general databank (EMBL, GenBank) contains a comprehensive and validated set of such protein sequences. After the creation of an identication engine for these proteins using pattern recognition tools, cross-validation and retro-validation of these proteins in even not completely annotated genomes, we up the first online database of ribosomal protein, Ribo-DB. The paper has been published in Molecular Biology and Evolution [Jauffrit et al., 2015].
The website uses classACNUC, a Python-class that I developed with Manolo Gouy to query the ribo-DB database but also other ACNUC database.
In the Ribo-DB team I am responsible for operating algorithms of the database, but also for the overall integration and the organization of servers. I am also in charge of the application face of the work and was the basis for the involvement of bioMérieux in the project (ANRT CIFRE PhD F. Jauffrit).
In addition to the development of this project and its deployment we can use its contents for scientific publications. We have ongoing reanalysis of evolutionary relationships of Actinobacteria (Figure below) and within this, the case of Mycobacteria wich is medically important.
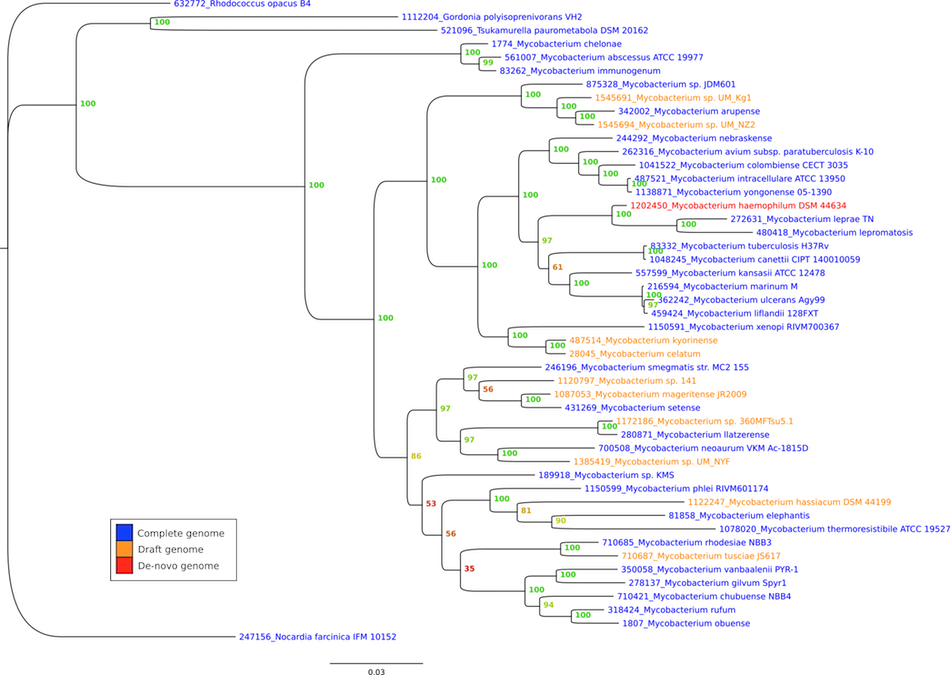
Understanding the emergence of the most pathogenic groups (Mycobacterium tuberculosis and Mycobacterium leprae) will be addressed with this new tool.
BIBI-DB
The BIBI project began in 2003 and was renewed in 2007-2008 and continuous improvements are made every year. It involves four researchers and an engineer.
Construction of the SSU-rDNA (16S-23S) database
The source of information concerning Type Strains sequences is now both SILVA and LPSN and the taxonomy DB is now the EMBL-ENA taxonomy.
The key point is the now the use of logical rules to give a relevant name to sequences sharing the same URS.
Construction of the CDS Database
ACNUC provides powerful and fast querying and retrieval from a variety of nucleotide and protein sequence databases, including EMBL, GenBank and RefSeq. The ACNUC system is designed to allow most fields of the sequence annotations to be used as entry points to the databases and combined in complex queries. These are elaborated using ACNUC query language that allows expression- and logical operator-based combination of retrieval criteria. Each query generates the list of all matching elements in the queried database.
The Python class classACNUC automates the query processing and merges relevant information extracted from the database sequence description data (taxonomy, strain, lengths, position etc) in a compact format in the `FASTA commentary line`. It also allows the filtering of the sequences according to length. The resulting files are thus more readily usable for further analysis. `classACNUC` relies on the `ACNUC Python API . See leBIBI-PPF site for an application. This python class was initially designed for the leBIBI-QBPP and riboDB database construction engines. A general-use version of `classAcnuc` is available upon request.
The databases
Several databases devoted to various markers are integrated in leBIBIQBPP. The largest one is for SSU rDNA. Others are smaller databases of general interest (rpoB) and databases that are relevant for a restricted spectrum of bacteria or for niche applications (e.g., sodA, groEL2). Note that other databases devoted to specific applications or research projects are also available upon request.
The SSU rDNA databases have five “flavors”:
- The “lax” database contains all bacterial and archaeal SSU rDNA sequences of GenBank that are identified at the genus level and lower (species, subspecies). It contains a large amount of not fully identified sequences. The coverage of geno-diversity is maximum in the “lax” database. The sequences that remains outside this "lax" DB are something like "dark matter".
- The “stringent” database contains sequences that are identified at the species level with a valid name according to the bacterial nomenclature. It also contains sequences of type strains of newly described bacteria or archaea, an indication that their names are under consideration for eventual validation. These two databases contain a lot of identical sequences and are rather frequently affected by erroneous species identifications.
- The “TS-stringent” database contains only reference sequences (either type strains or, if missing, algorithm-chosen), so that newly described or non validly published species may be missing. Identification errors are almost absent.
- Lastly, the “genus-level” database is a subset of the “TS-stringent” database containing only one sequence for each genus: the sequence of the TS of the genus type-species.
The major advantage is that the databases are all subsets of the database "lax" and combining a very restrictive level (genusonly) and a wider level (TS-stringent) makes a representation at a large evolutionary scale while allowing also a high resolution around a group of interest.
Coupled with the leBIBI-QBPP query tool such databases enable to optimize the phylogenetic position of an unknown sequence.