Ribo-DB
Le projet Ribo-DB est des projets phares de notre équipe, il implique 4 enseignants-chercheurs ou chercheurs, 2 ingénieurs et un doctorant.
Les protéines ribosomiques sont de plus en plus utilisées car leur nombre (90 protéines), la faible probabilité de transfert horizontaux, leurs caractéristiques évolutives en font des outils indispensables pour l'étude de l'évolution des organismes (phylogénie à large échelle ou à courte échelle évolutive). Par ailleurs ces protéines sont utilisées pour l'identidication en routine des bactéries pathogènes à l'aide de la spectrométrie de masse MALDI-TOF et sont aussi des cibles potentielles pour l'amplication par PCR dans des outils diagnostiques.
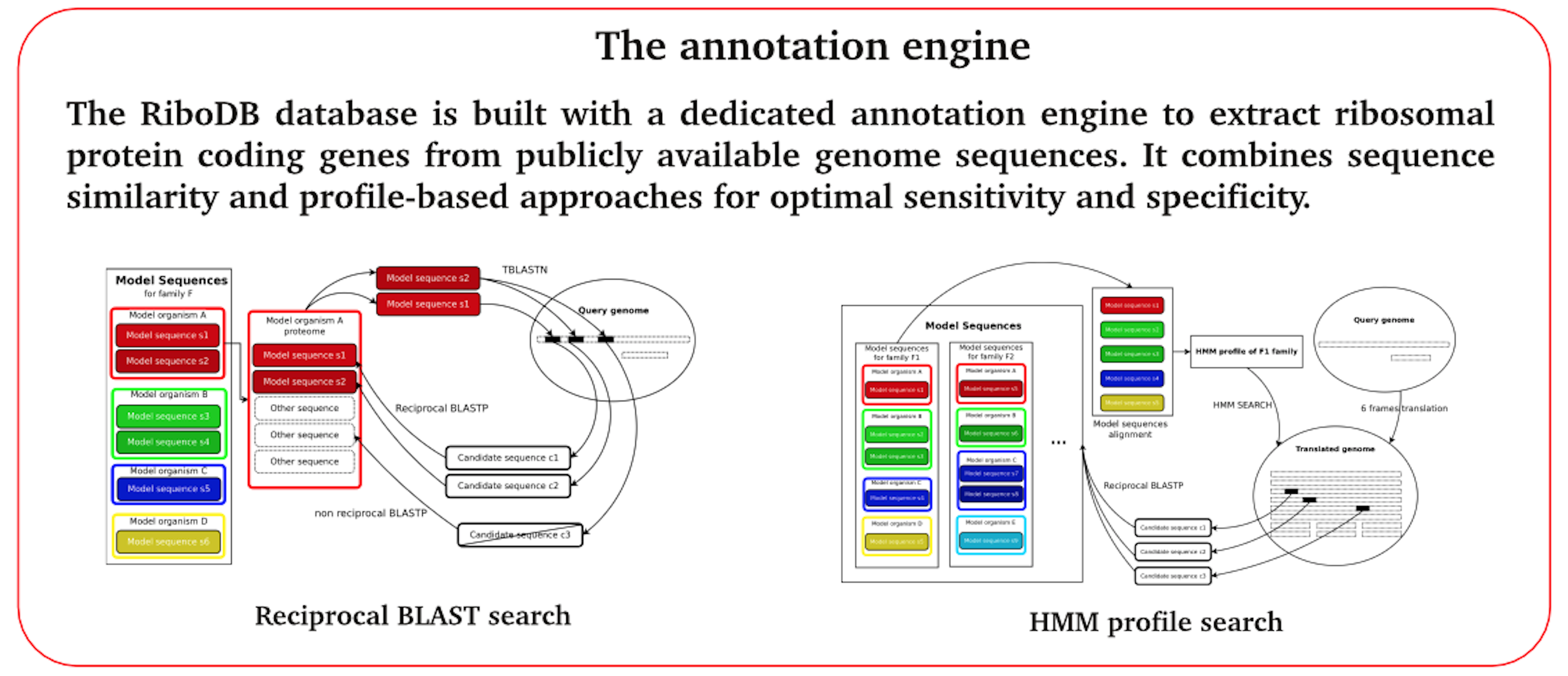
Aucune base de donnée généraliste (EMBL, GenBank) ne contient un ensemble exhaustif et validé des séquences de protéines. Après avoir développéun moteur d'identication de ces protéines utilisant des outils de reconnaissance de profil, des validations croisées et des rétro-validations de ces protéinesdans des génomes complets même non annotés, nous avons donc mis en ligne lapremière base de donnée de protéines des ribosomes au niveau mondial, Ribo-DB . L'article publié dans Molecular Biology and Evolution [Jauffrit et al., 2015].
Ce site utilise classACNUC, classe écrite en langage Python que j'ai développée avec Manolo Gouy pour interroger la base de séquences ribo-DB mais aussi les bases de données ACNUC de séquences de façon générique.
Dans l'équipe Ribo-DB je suis chargé des algorithmes d'exploitation de la base de données, mais aussi chargé de l'intégration générale et de l'organisation des serveurs. Je suis aussi en charge de la face applicative du projet et j'ai été à la base de l'implication de la société bioMérieux dans le projet (ANRT, Doctorat CIFRE de F. Jauffrit).
Outre le développement de ce projet et son déploiement nous allons pouvoir exploiter son contenu par des publications scientifiques. Nous avons en cours la réanalyse des relations évolutives des Actinobactéries (Figure ci-dessous) et au sein de cet ensemble du cas des Mycobactéries qui est médicalement important.
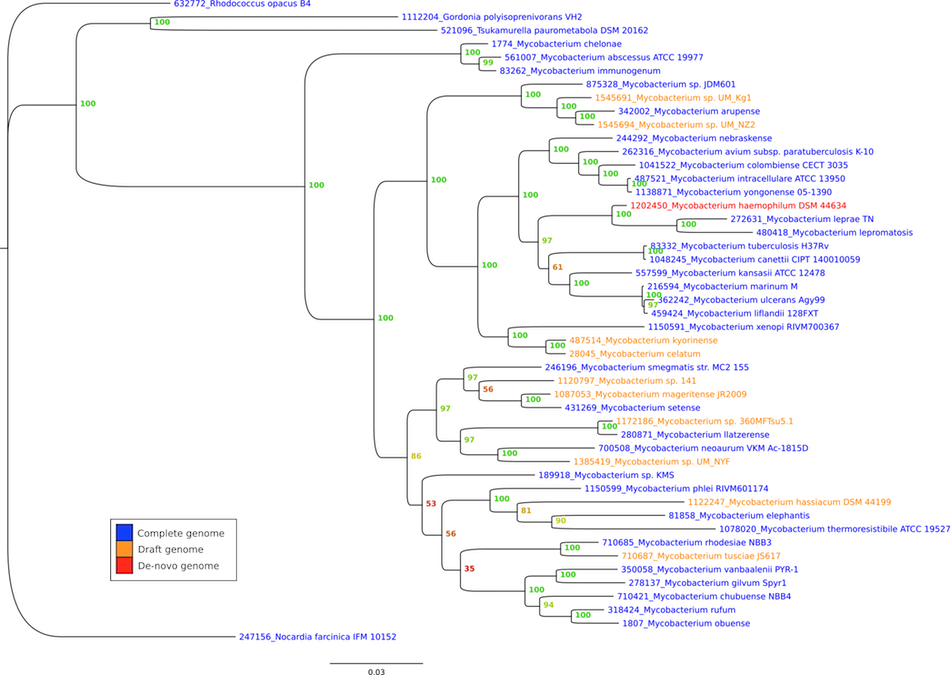
La compréhension de l'émergence des groupes les plus pathogènes (Mycobacterium tuberculosis et Mycobacterium leprae) va être abordé avec ce nouvel outil.
BIBI-DB
le projet BIBI a été initié en 2007-2008 est poursuivi et des améliorations constantes sont apportées. Il implique 3 enseignants-chercheurs ou chercheurs et un ingénieur.
Principe de construction
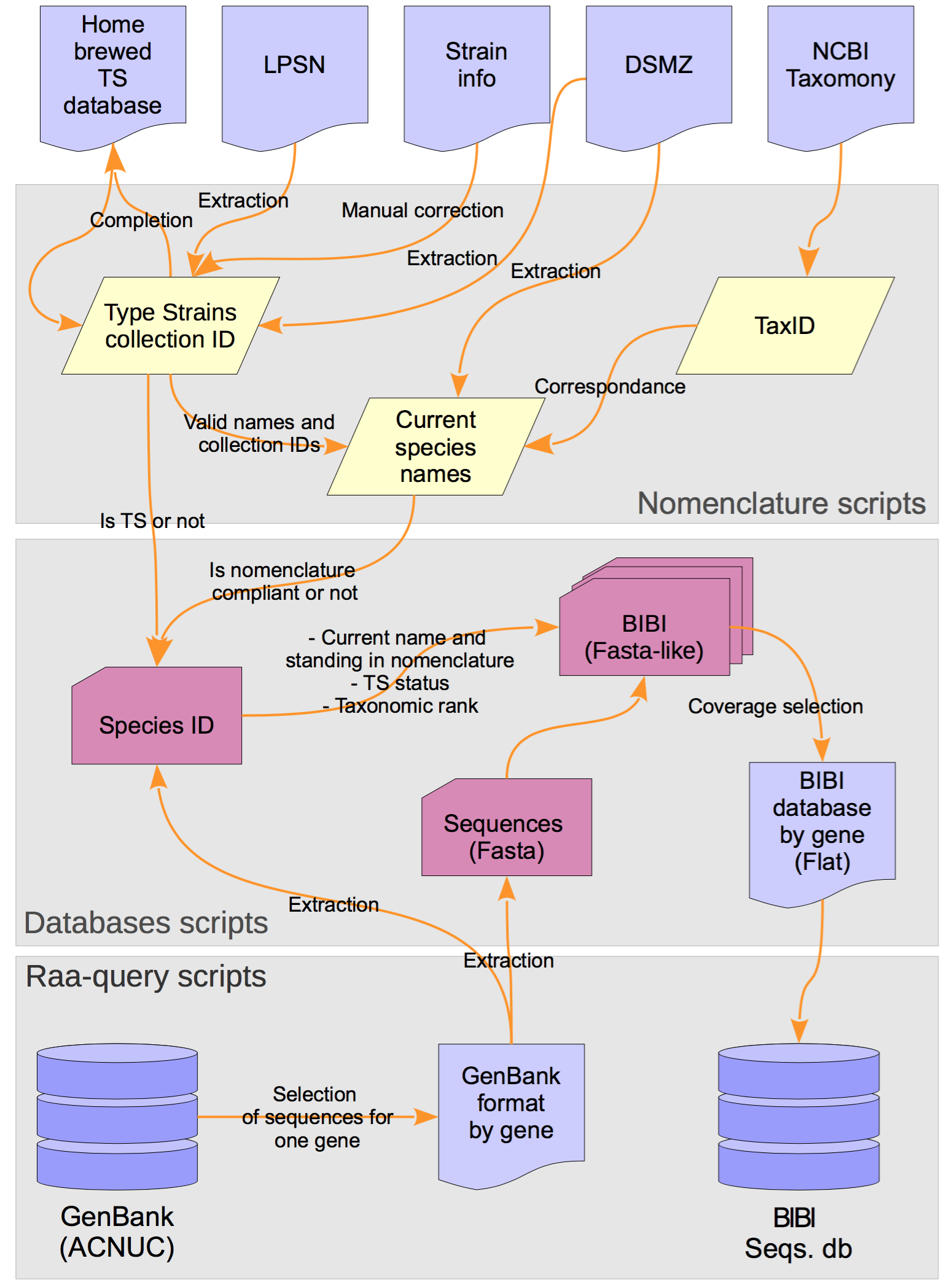
Les bases de données
Plusieurs bases de données consacrées à divers gènes sont intégrés dans BIBI-DB. Le plus important est le SSU-rDNA. D'autres sont des bases de données plus petites d'intérêt général (rpoB) et des bases de données qui sont pertinentes pour un spectre restreint de bactéries ou pour des applications de niche (par exemple, sodA, groEL2). Notez que d'autres bases de données consacrées à des applications ou des projets de recherche spécifiques sont également disponibles sur demande.
Les bases de données SSU ADNr ont cinq «saveurs» :
- La base de données «lax» contient toutes les séquences SSU ADNr bactériennes et des archées de GenBank, sauf ceux pour lesquels aucune information taxonomique plus spécifique que Bacteria ou Archaea est rapporté. Il est très complet, mais il contient une grande quantité de séquences none complètement identifiés. La couverture des genovars est maximale dans la base de données «lax».
- La base de données "stringent" contient des séquences qui sont identifiées au niveau de l'espèce un nom valide selon la nomenclature bactérienne. Il contient également des séquences de souches de type de bactéries ou archées nouvellement décrites, une indication que leurs noms sont à l'étude pour la validation éventuelle. Ces deux bases de données contiennent un grand nombre de séquences identiques et sont assez fréquemment affectés par des identifications erronées d'espèces.
- La base de données "TS-stringent" ne contient que des séquences de souches de type (TS), de sorte que nouvellement décrit ou non valablement publiée espèces peuvent être manquantes. Cette base de données est moins susceptible d'être contaminé par des identifications erronées d'espèces.
- La base de données "superstringent" est un sous-ensemble de la précédente où une seule ou un petit nombre de séquences est conservée pour chaque espèce. Les séquences sont ceux marqués dans la liste des procaryotes noms avec permanent dans la Nomenclature (LPSN) comme séquence de référence pour une espèce donnée. erreurs d'identification sont presque absents, mais les espèces nouvellement décrites ou des espèces valablement publiée non sont souvent absents.
- Enfin, la base de données "au niveau du genre" (genuslevel) est un sous-ensemble de la base de données "superstringent" contenant une seule séquence pour chaque genre: la séquence du TS du genre espèce-type.
L'intérêt majeur est que les bases de données sont toutes des sous-ensembles de la base "lax" et que combiner un niveau très restrictif (genusonly) et un niveau plus large (TS-stringent) permet une représentation de la phylogénie à grande échelle évolutive tout en permettant autout d'un groupe d'intérêt une grande résolution.
Couplé à l'outil d'interrogation leBIBI-QBPP ces bases permettent d'optimiser le positionnement phylogénétique d'une séquence inconnue.