Ribo-DB
Site Web
L'interface web permet de construire une interrogation complexe et d'extraire les séquences correspondantes.
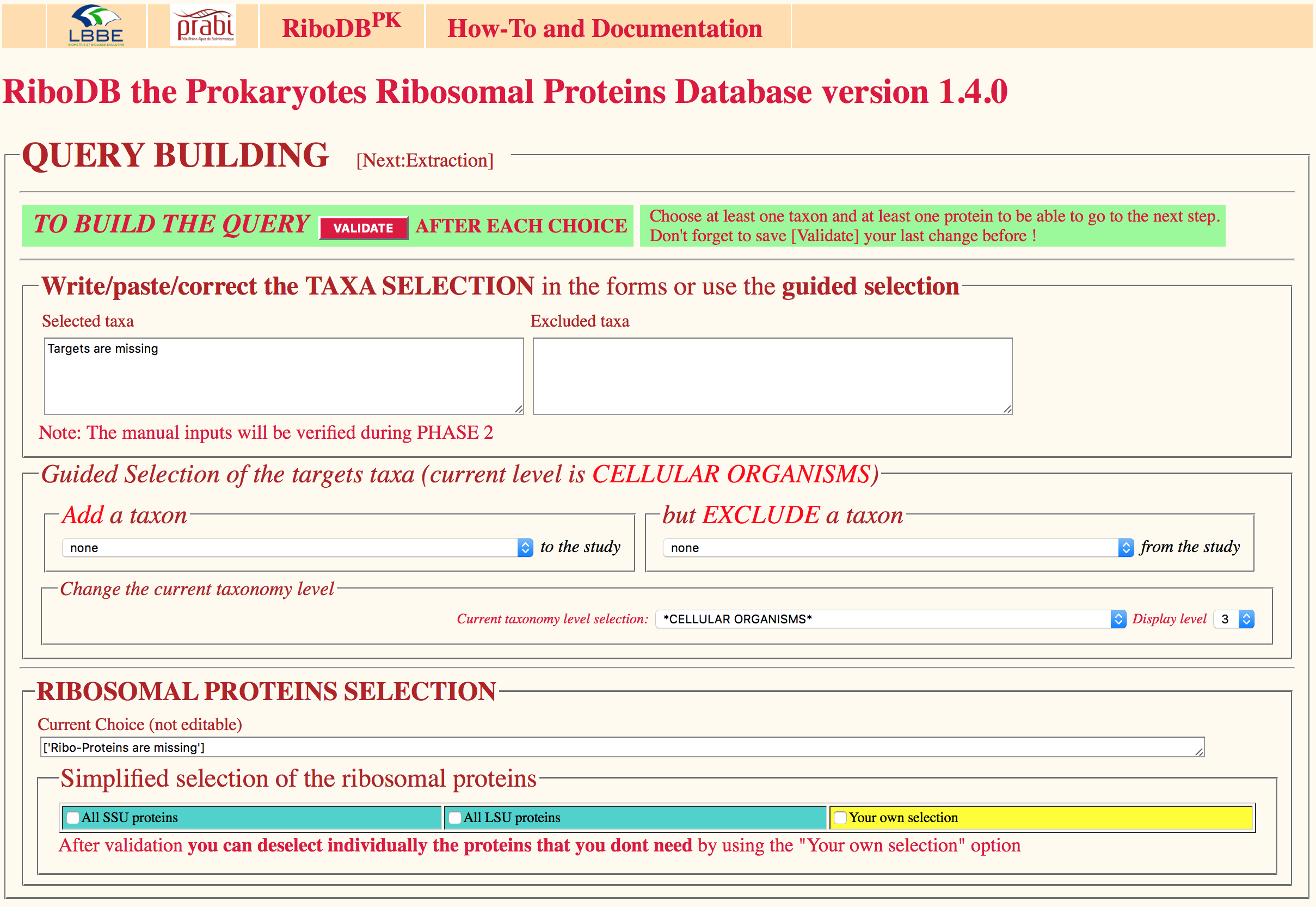
L'interface web à RiboDB permet aux utilisateurs d'extraire des séquences nucléotidiques et/ou protéiques des r-protéines pour un ensemble donné de souches. La sélection d'un niveau taxonomique parmi les archées et bactéries est possible, ainsi que toute combinaison ou exclusion des niveaux taxonomiques. La classe Python ClassACNUC permet de composer une interrogation complexe (depuis un script ou la ligne de commande) et d'extraire les séquences correspondantes.
La navigation dans l'arborescence de la taxonomie des procaryote est possible avant la sélection, mais les niveaux de taxonomie désirés peut être aussi soumis directement dans l'interface. Ce navigateur de taxonomie repose sur un serveur de taxonomie ACNUC lié à la base de données RiboDB.
La sélection de l'une/plusieurs des protéines ribosomiques parmi 90, ou d'un sous-ensemble donné (petite ou grande sous-unité du ribosome), voire de la totalité est directement possible. Une page de résultat montre l'évolution des requêtes et du processus d'extraction.
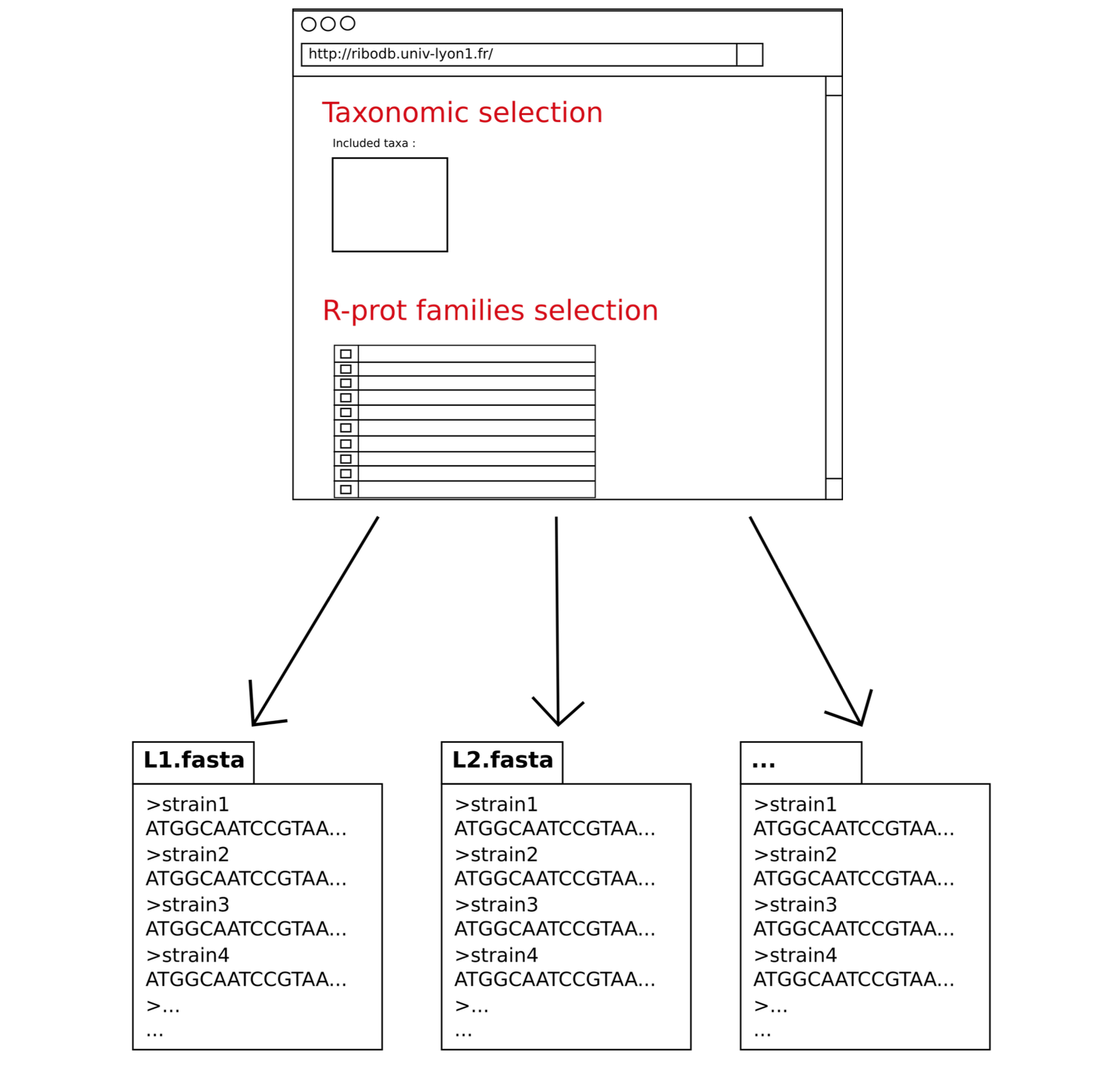
Le résultat final est un ensemble de fichiers FASTA (séquences nucléotidiques et protéiques) correspondant aux protéines ribosomiques sélectionnées et organisées dans des répertoires. Les répertoires ou fichiers peuvent être consultés et téléchargée
 Les ensembles de données pour le débarassé des paralogues sont également disponibles pour une utilisation directe dans les programmes de phylogénie. Une archive compactée est également disponible.
La ligne de commentaire FASTA condense tous les principaux descripteurs d'une séquence de protéine ribosomique (espèce, souche, assemblyID, BankID, la position, brin, hiérarchie taxonomique) d'une manière structurée.
Les ensembles de données pour le débarassé des paralogues sont également disponibles pour une utilisation directe dans les programmes de phylogénie. Une archive compactée est également disponible.
La ligne de commentaire FASTA condense tous les principaux descripteurs d'une séquence de protéine ribosomique (espèce, souche, assemblyID, BankID, la position, brin, hiérarchie taxonomique) d'une manière structurée.
Le site est organisé comme un système client-serveur classique qui utilise python et des CGI-python pour communiquer. La classe python classACNUC gère l'interface de requête à ACNUC et les extractions.
ClassACNUC
ClassACNUC est une classe Python qui a été développé initialement pour le projet riboDB. Elle se fonde sur le potentiel du python ACNUC API. L'utilisation de ClassACNUC est décrite dans son site
Cette classe python essaie de simplifier l'écriture des requêtes et la création des listes correspondantes en automatisant une grande partie du processus sous-jacent. Cette classe python modifie également le processus d'extraction de séquence et permet de fusionner les informations souhaitées extraites de la base de données (taxonomie, les souches, les longueurs, la position Etc.) sous une forme compacte dans une ligne de commentaire de FASTA. Le contenu de la ligne de commentaire FASTA est ainsi définie par l'utilisateur.
Les fichiers résultants sont donc rapidement utilisables pour d'autres analyses, car les informations pertinentes pour la phylogénie et de contrôle de la qualité sont directement accessibles.
Une requête en ligne de commande, très simplifiée est disponible.
L'outil d'interrogation leBIBI-QBPP permet la soumission de séquences inconnues et l'obtention automatique du positionnement phylogénétique.BIBI-DB interface web
Le processus d'analyse
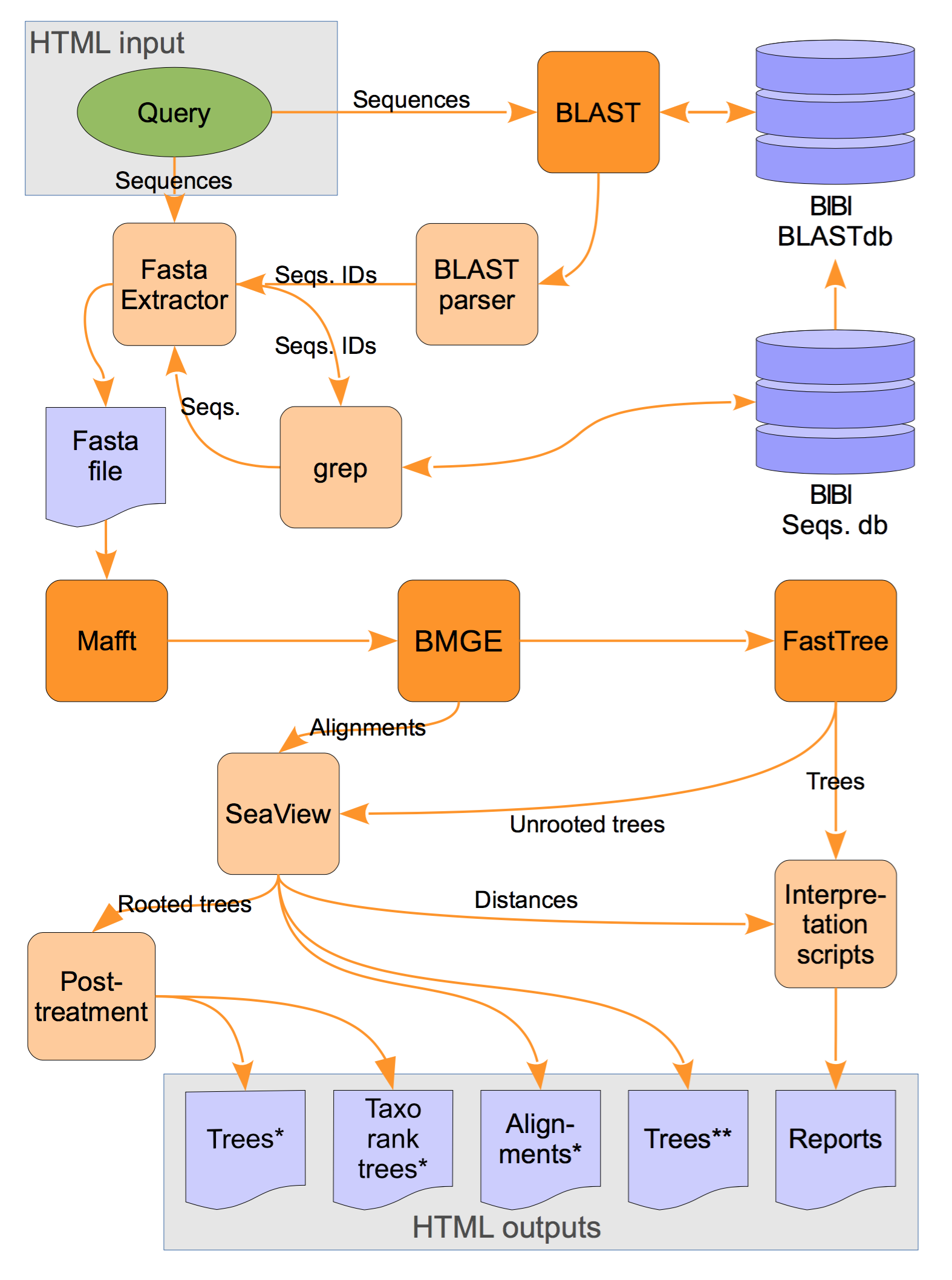
Organisation du pipeline leBIBI-QBPP. BLASTN est exécuté avec requête sur la base de données sélectionnée. Un ensemble de séquences présentant les scores les plus élevés de similarité est extrait de la base de données. Les séquences sélectionnées sont alignées par mafft le programme BMGE est utilisé pour optimiser l'alignement des séquences et FastTree est ensuite utilisé pour reconstruire l'arbre par maximum de vraisemblance approché. Les scripts Python analysent les résultats et l'organisation des sorties graphiques est générée par SeaView (voir en bas de page pour les liens de logiciels).
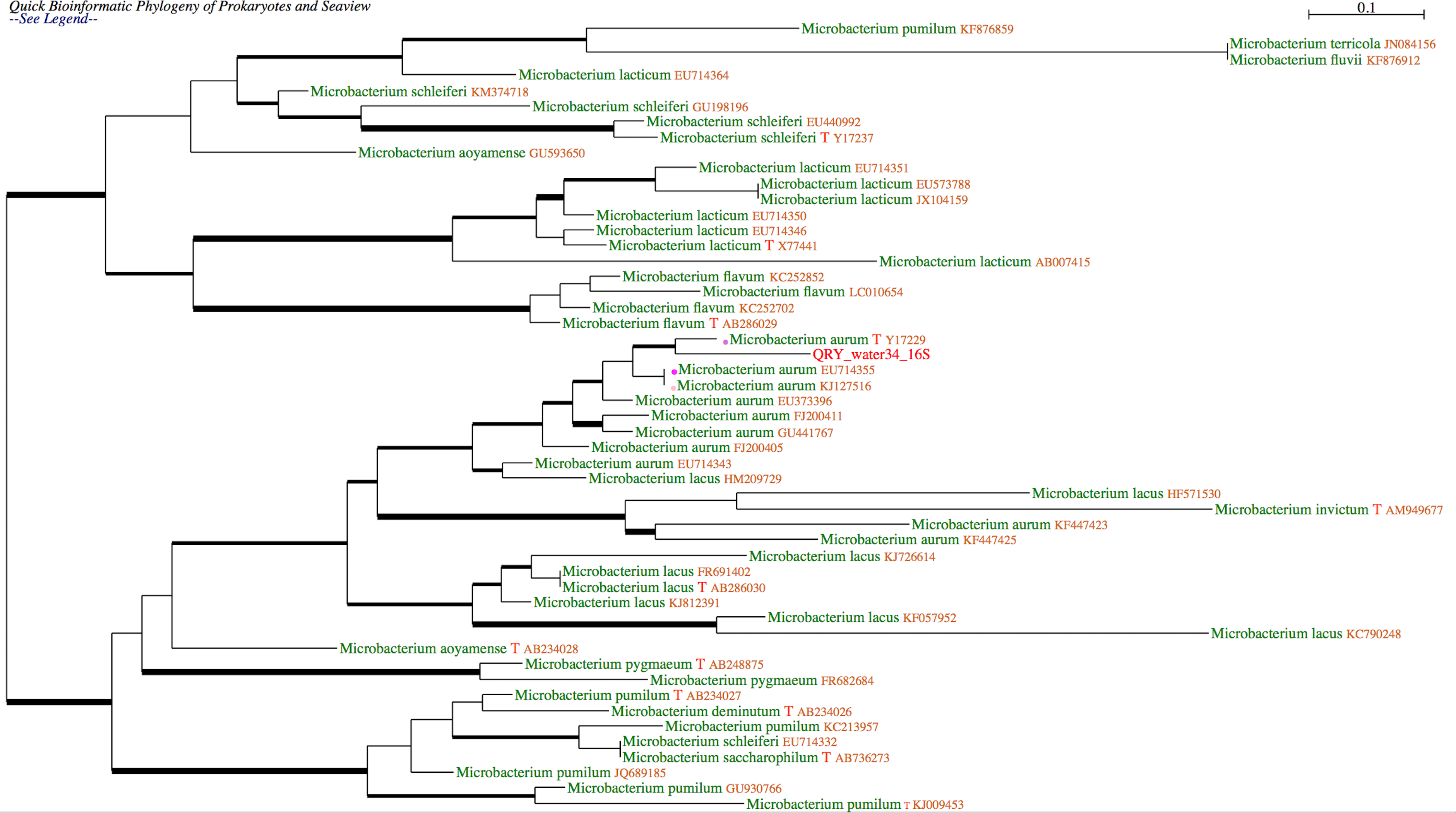
Exemple d'arbre
Placement phylogénétique d'une séquence bactérienne. La séquence d'interrogation est soupçonnée être une espèce inconneue de Mycobacterium. Ceci a été confirmé par l'analyse de la séquence rpoB obtenue à partir du même extrait bactérien et la base rpoB "stringent"